
coming soon:





Drug-target 3D structure predictions and binding affinity
An open-source protein binding affinity prediction tool developed by researchers at MIT in collaboration with Recursion
Small molecule binding affinity, or the strength of the interaction between a protein and a chemical ligand, plays a crucial role in the function of small molecule drugs. Binding affinity prediction is critical for molecule screening — but something that no AI model has been able to accurately predict previously.
Running these experiments in the lab is a critical time and cost bottleneck for early stage drug discovery. The best computational techniques are atomistic, long physics-based simulations like free-energy perturbations (FEP) which have high accuracy but are far too slow and expensive to be used extensively for small molecule discovery.

Boltz-2 is a new biomolecular foundation model that jointly predicts structure and binding affinity.
Delivering accurate binding calculations a thousand times faster than free energy perturbation, the current gold standard.
A New Era in Biomolecular Interaction Modeling
The accuracy and speed of Boltz-2 allows large scale virtual screening with significantly higher enrichment factors than previous methods — improving on the structural accuracy of Boltz-1 complexes by expanding the training data with molecular dynamics ensembles and self-distilled predictions from Boltz-1.
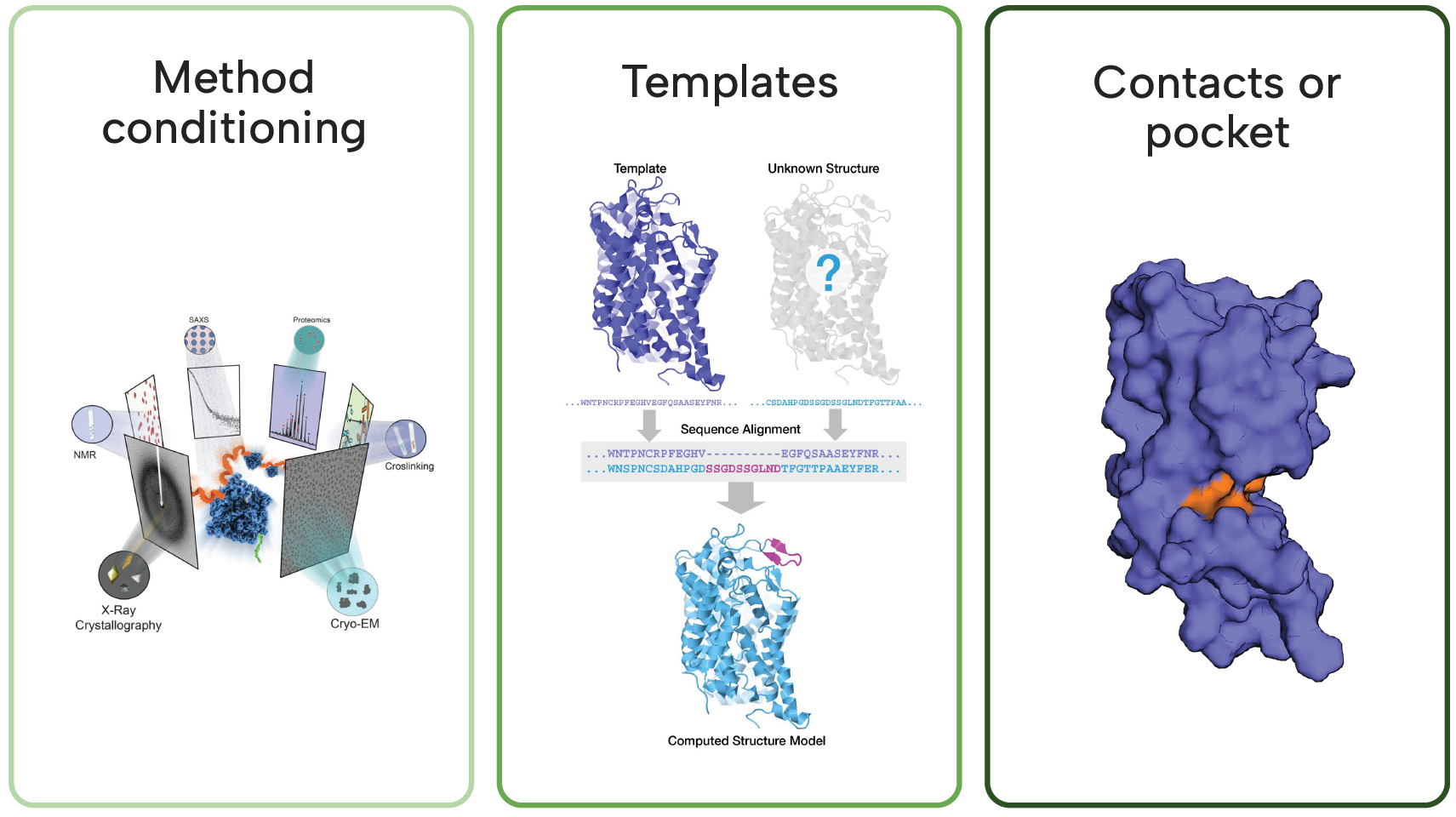
Additionally, Boltz-2 includes several features that make the model more controllable with experimental data or human intuition, making it more practically useful for drug development to serve as the basis of a next generation of pipelines. These features include:
- Method conditioning — allows users to specify an experimental modality to emulate
- Template steering — allows users to input reference templates that embed prior knowledge
- Contact/pocket constraints — ensure output follows respects given conditions
This model can be used to do virtual screening — hit discovery and hit to lead — providing meaningful improvement over the state-of-the-art on unseen targets, without any fine tuning.
Boltz-2 Affinity, a protein-ligand affinity model trained on millions of batch-corrected assay values aggregated from the literature, demonstrates unprecedented binding affinity as the first deep learning model to approach the accuracy of physics-based free-energy perturbation (FEP) methods, while running 1000x faster — making highly-accurate in silico screening practical for early-stage drug discovery.
This is the first deep learning model to get close to FEP performance, representing a significant step forward for computational chemistry and small molecule design.
Boltz-2 Access
The Boltz-2 model code is open-sourced under a MIT license permitting academic and commercial use.
The model, weights, and training pipeline are available on Github.

